General remarks on WIEN2k
WIEN2k consists of many independent Fortran90 programs, which are linked together via C-shell scripts. You can run WIEN2k using any www-browser and the w2web interface, but of course more experienced users can run WIEN2k also from the command line.
The main tasks are:
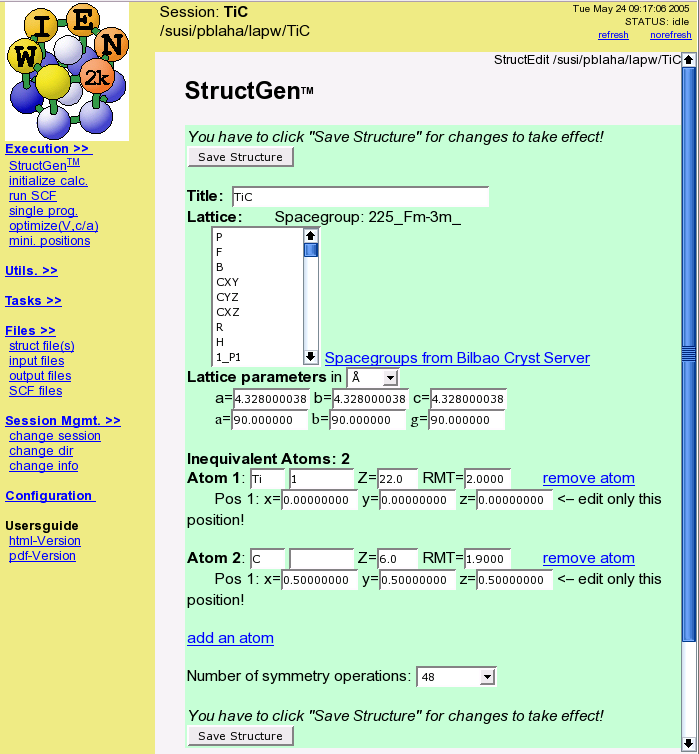
- Define your structure (cif-file import, spacegroup support, symmetry detection)
- initialize (semi-automatic guided input generation)
- run scf-cycle (with/without simultaneous optimization of atomic positions)
- Calculate some properties ("Guided Tasks" in w2web)
- write a publication (NOT yet supported in w2web, you must do it yourself)
{kind=link}
{kind=link}
Features and Calculated Properties
- LDA, GGA, meta-GGA (libxc interface), LDA+U and EECE, orbital polarization, Hybrid-DFT
- centro- or non-centrosymmetric cells (mode), all 230 spacegroups built in
- spin-polarization (ferro- or antiferromagnetic structures), spin-orbit coupling
- sequential mode, k-parallel mode (without MPI, slow network with common NFS), massively parallel MPI mode (shared memory or Infiniband)
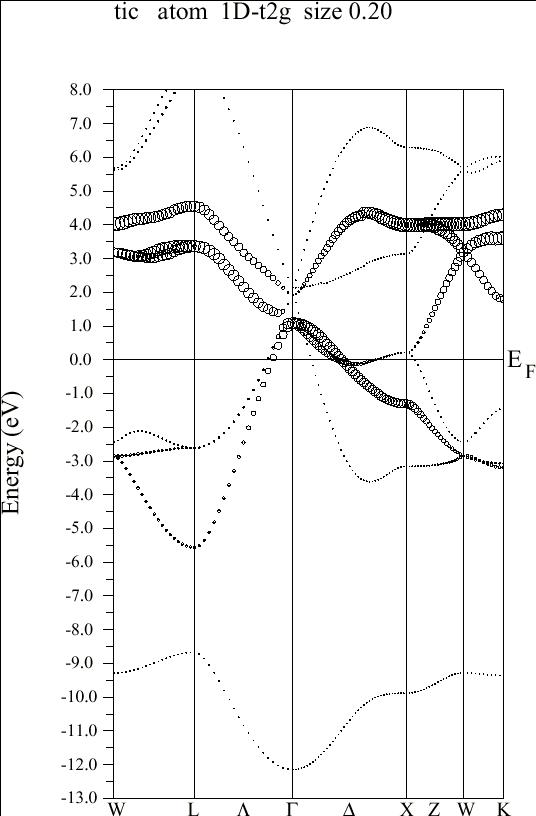
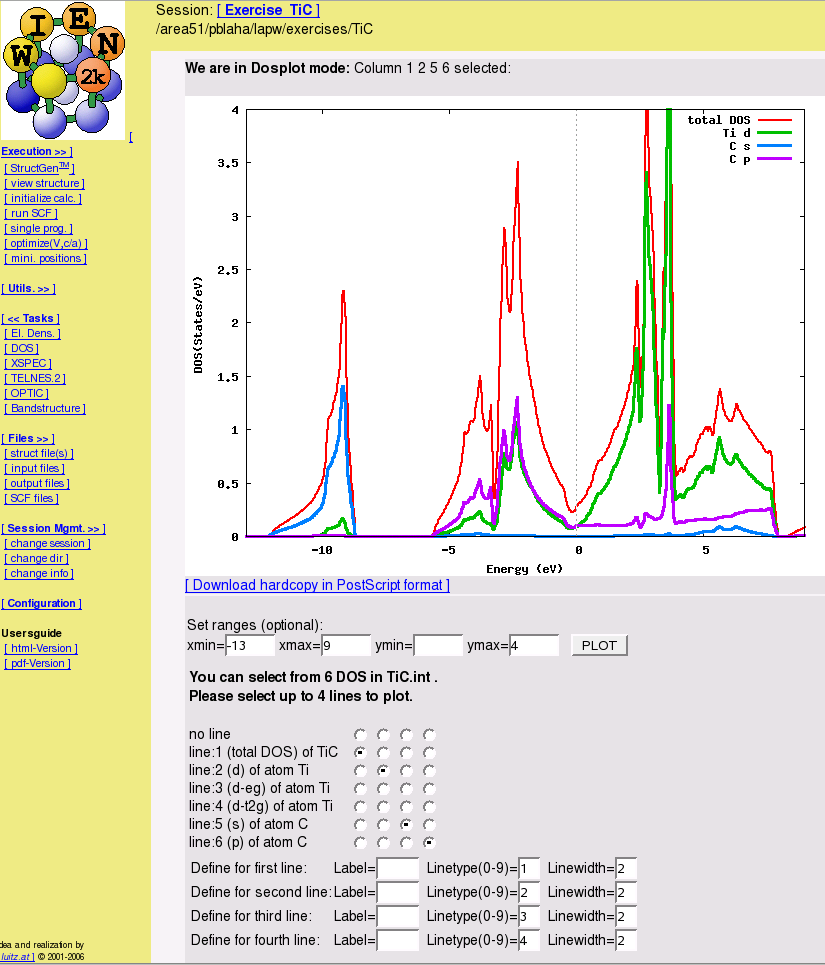
- Energy bands and density of states
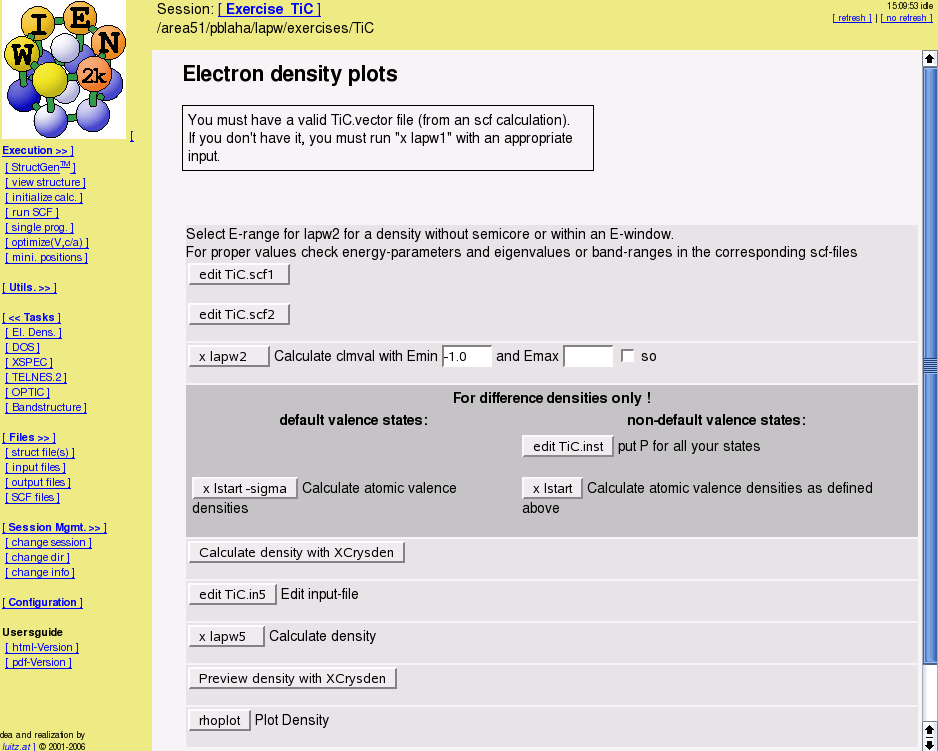
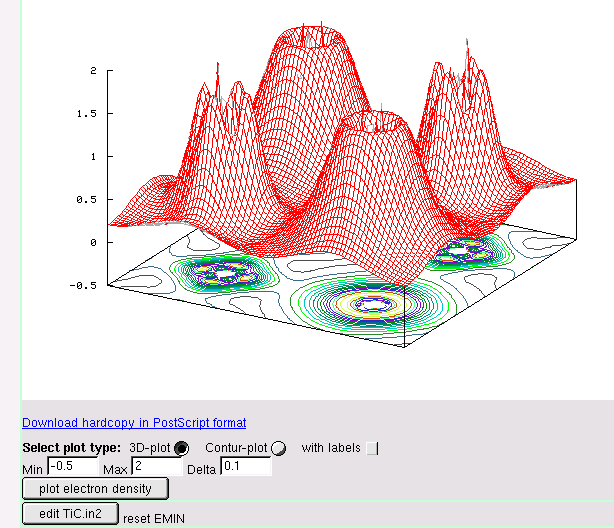
- electron densities and spin densities, x-ray structure factors, potentials, STM and AFM simulations
- Baders's "atoms-in-molecule" concept
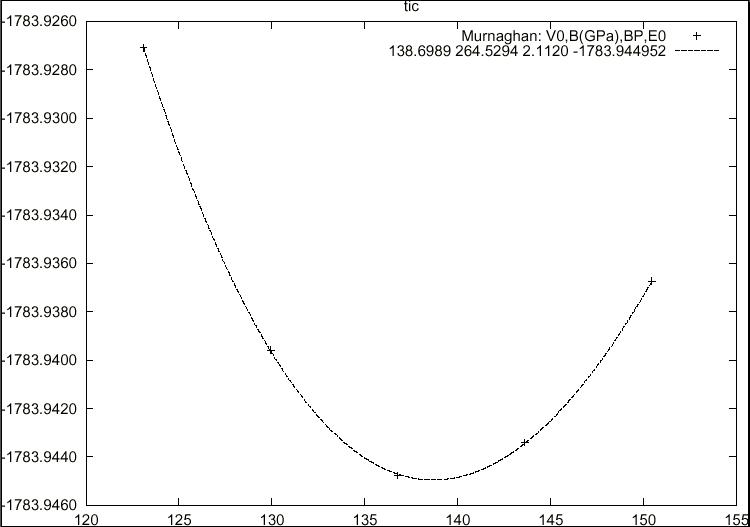
- total energy, forces, equilibrium geometries, structure optimization, elastic constants, molecular dynamics
- Phonons, with an interface to K.Parlinski's PHONON or A. Togos Phonopy program
- electric field gradients, isomer shifts, hyperfine fields, NMR chemical shifts, NMR Knight shifts
- x-ray emission and absorption spectra, electron energy loss spectra
- optical properties
- fermi surfaces
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A very recent extensive overview is given in:
WIEN2k: An APW+lo program for calculating the properties of solids.
P. Blaha, K.Schwarz, F. Tran, R. Laskowski, G.K.H. Madsen and L.D. Marks,
J. Chem. Phys. 152, 074101 (2020)
©2001 by P. Blaha and K. Schwarz